INTESTINAL VILLI WITH BACTERIA
Microbiome Fundamentals
Microbiome Fundamentals

Measuring The Microbiomes
Historically, identification of microorganisms relied on culture-based methods. This method has significant limitations, including an inability to culture all organisms in the microbiome.1-3
Not all microorganisms are viable in the in vitro conditions associated with culture-based methods.1
Next-generation molecular methods use bacterial DNA sequences as proxies for the organisms to identify taxonomy, relative abundance, and function.1,4-6 The most commonly used methods are briefly described below.
Overview of commonly used methods for measuring the microbiome
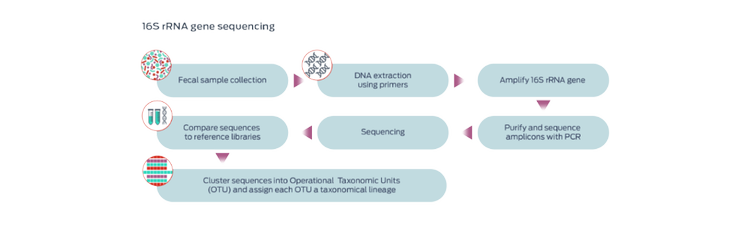
Amplicon-based marker gene sequencing
The most commonly used approach involves sequencing the 16S ribosomal RNA (rRNA) gene, present in all bacteria and archaea but not present in fungi or the animal kingdom, to generate a microbial census of the sample.1,4,7 Identification of the mycobiome substitutes 18S rRNA genes.7
The 16S rRNA gene is highly conserved across bacterial species, and includes hypervariable regions that allow differentiation.1 This approach allows identification of many microorganisms from a sample, including microorganisms that cannot be successfully cultured.1 However, this technique does not distinguish between microorganisms that are live or dead, transient or resident, commensal or pathogenic, or resistant or sensitive to antibiotics.1
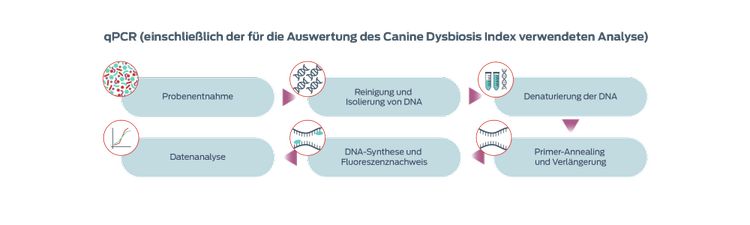
Quantitative polymerase chain reaction (qPCR)
Quantitative PCR is a quick and affordable method of quantifying specific bacterial taxa of interest, and allows the development of reference intervals. Based on qPCR for seven bacterial species, a Canine Microbiota Dysbiosis Index has been developed to track changes in the microbiome in response to therapy.3
Describing the "microbial census"
Operational Taxonomic Units (OTUs) are clusters of the DNA sequences based on similarity. Representative sequences from each OTU are compared to reference databases to assign identification of the microbes in the cluster.
Alpha diversity and beta diversity indicate the range of species in the microbiome. The alpha diversity is a measure of the diversity within a sample.1 Alpha diversity takes into account the richness (the number of species/OTUs and their distribution) and the evenness (the relative abundance of the different OTUs/species and their distribution). Alpha diversity is based on algorithms and is commonly expressed as an index such as Shannon’s index or Simpson’s index.1 Beta diversity is a measure of the dissimilarity between samples, often represented as cluster patterns. Systems used to calculate the beta diversity include Bray-Curtis and the Unweighted or Weighted UniFrac.1
Expanding from census to function
As research continues, two main questions arise with regard to the microbiomes: what microbes are present, and what are they doing? Identification of the microorganisms present in the microbiome does not provide information regarding their function.3,5 It is likely that certain metabolic and molecular functions are performed by more than one microbe, resulting in a redundant ecosystem to provide flexibility and resiliency.3,8
Due to this redundancy, analysis of the microbiome based on the microbial species present in the population is insufficient for the detection of functional changes in the microbiome. Although the relative abundance of the microbes may or may not be altered by an intervention or condition, the microbiome present may shift microbial activities and metabolism to compensate; in order to detect these shifts, different analytical procedures are necessary.
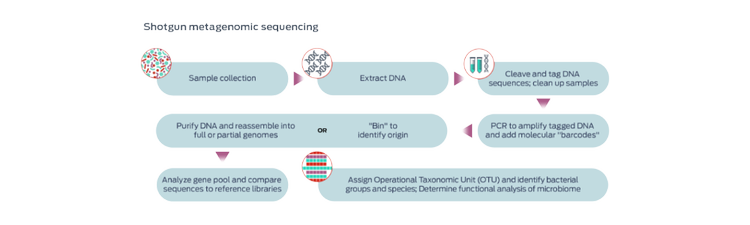
Shotgun metagenomic sequencing
Shotgun methods, so named because they are untargeted (not intended to detect the presence of a predetermined set of microbes), are gaining popularity in pet microbiome analysis. These methods offer the advantage of sequencing functional genes, as opposed to simply identifying bacterial identification.3-5 However, these processes require larger amounts of DNA from samples and are more expensive.3
Long-read sequencing facilitates the assembly of complete genomes, including genes usually missed with short-read metagenomics and which provide additional biological insights.6
Metabolomics, proteomics and transcriptomics
These processes measure the functional activity in a microbiome.4 Metabolomics approaches are used to determine the metabolite profile, usually characterized through processes such as nuclear magnetic resonance, spectroscopy, mass spectroscopy and liquid chromatography.7 These approaches investigate pathways regulated by microbes, such as short-chain fatty acid production, bile acid metabolism, neutrotransmitter production, and indole production.3

Explore other areas of the Microbiome Forum



Find out more
- Belas, A., Marques, C., & Pomba, C. (2020). The gut microbiome and antimicrobial resistance in companion animals. In Duarte, A. & Lopes da Costa, L. (Eds.), Advances in Animal Health, Medicine and Production (1st ed.), pp. 233—245. Springer International Publishing
- Cunningham, M., Azcarate-Peril, M. A., Barnard, A., Benoit, V., Grimaldi, R., Guyonnet, D.,…Gibson, G. R. (2021). Shaping the future of probiotics and prebiotics. Trends in Microbiology, 29(8), 667—685. doi:10.1016/j.tim.2021.01.003
- Pilla, R., & Suchodolski, J. S. (2021). The gut microbiome of dogs and cats, and the influence of diet. Veterinary Clinics of North America Small Animal Practice, 51(3), 605–621. doi:10.1016/j.cvsm.2021.01.002
- Bokulich, N. A., Ziemski, M., Robeson, M. S., & Kaehler, B. D. (2020). Measuring the microbiome: Best practices for developing and benchmarking microbiomics methods. Computational and Structural Biotechnology Journal, 18, 4048–4062. doi:10.1016/j.csbj.2020.11.049
- Radjabzadeh, D., Uitterlinden, A. G., & Kraaij, R. (2017). Microbiome measurement: Possibilities and pitfalls. Best Practice & Research Clinical Gastroenterology, 31, 619–623. doi:10.1016/j.bpg.2017.10.008
- Cusco, A., Pérez, D., Viñes, J., Fàbregas, N., & Francino, O. (2020). Long-read metagenomics retrieve complete single-contig bacterial genomes from canine feces. In review, BMC Genomics. doi:10.21203/rs.3.rs-135952/v1
- Marchesi, J. R. & Ravel, J. (2015). The vocabulary of microbiome research: a proposal. Microbiome, 3, 31. doi:10.1186/s40168-015-0095-5
- Koidl, L., & Untersmayr, E. (2021). The clinical implications of the microbiome in the development of allergy diseases. Expert Review of Clinical Immunology, 17, 115—126. doi:10.1080/1744666X.2021.1874353